|
1999年 9 月作成 規制区分: 劇薬、指定医薬品、要指示医薬品 (注意−医師等の処方せん・ 指示により使用すること) 貯法:室温保存 使用期限:包装に表示 |
抗ウイルス化学療法剤 (硫酸アバカビル製剤) |
 |
| |
|
| 【組成・性状】 | |
| 1. | 組成 本剤は、1錠中に硫酸アバカビル351mg(アバカビルとして300mg)を含有する。 |
| 2. | 性状 本剤は黄色のフィルムコート錠である。 |
| 販売名 | 識別コード | 表 (直径) |
裏 | 側面 (厚さ) |
重量 |
| ザイアジェン錠 | GX 623 | 長径:約18.4mm 短径:約 7.2mm |
約6.0mm |
約814mg |
| 【効能・効果】 HIV感染症 | |||||||
| |||||||
| 【用法・用量】 通常、成人には他の抗HIV薬と併用し、アバカビルとして1回300mgを1日2回経口投与する。なお、年齢、体重、症状により適宜増減する。 | |||||||
| |||||||
| 【使用上の注意】 | ||
| 1. | 慎重投与(次の患者には慎重に投与すること) | |
| (1) | 腎障害患者[本剤の約2%が腎から未変化体として尿中に排泄される。腎障害患者における薬物動態学的データはなく、腎障害の程度に応じた推奨投与量は明らかでない。] | |
| (2) | 肝障害患者[本剤は主に肝で代謝される。非代償性の慢性肝疾患を有する患者において、有意な変化を認めたとの報告があるが、詳細は不明であり、肝障害のある患者への推奨投与量は明らかでない。なお、現在、肝障害患者における薬物動態試験が実施中である。] | |
| (3) | 高齢者(「高齢者への投与」の項参照) | |
| (4) | 妊婦又は妊娠している可能性のある婦人(「妊婦、産婦、授乳婦等への投与」の項参照) | |
| 2. | 重要な基本的注意 | ||
| (1) | 本剤はHIV感染症治療の経験を有する医師が投与を行うこと。 | ||
| (2) | 本剤投与による過敏症は、通常、治療開始6週以内(平均11日)に発現し、まれに致死的となることが報告されている。患者に過敏症について必ず説明し、過敏症を注意するカードを常に携帯するよう指示すること。過敏症の徴候又は症状が発現した場合は、直ちに担当医に報告させ、本剤の服用を中止すべきか否かを患者に指示すること。(「副作用」の項参照) | ||
| (3) | 過敏症の発現後は、本剤を決して再投与しないこと。本剤の再投与により数時間以内にさらに重篤な症状の発現、生命を脅かす程度の血圧低下の発現及び死に至る可能性がある。(「副作用」の項参照) また、過敏症が発現した患者には、本剤を二度と服用しないよう十分指導するとともに、担当医又は医療施設を変わる場合には本剤による過敏症が発現した旨を新しい担当医に伝えるよう十分指導すること。 | ||
| (4) | 本剤の使用に際しては、患者又はそれに代わる適切な者に、次の事項についてよく説明し同意を得た後、使用すること。 | ||
| 1) | 現時点では、本剤の薬物動態及び有効性・安全性は外国人の成績しかなく、日本人での臨床試験を実施し薬剤に関する科学的なデータを収集中であること。 | ||
| 2) | 本剤はHIV感染症の根治療法薬ではないことから、日和見感染症を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので、本剤投与開始後の身体状況の変化については、すべて担当医に報告すること。 | ||
| 3) | 本剤の投与後過敏症が発現し、まれに致死的となることが報告されている。過敏症を注意するカードに記載されている徴候又は症状(発熱、皮疹、疲労感及び嘔気、嘔吐、下痢、腹痛等の胃腸症状を含む)が発現した場合は、直ちに担当医に報告し、本剤の服用を中止すべきか否か指示を受けること。また、過敏症を注意するカードは常に携帯すること。 | ||
| 4) | 本剤を含む現在の抗HIV療法が、性的接触又は血液汚染を介した他者へのHIV感染の危険性を低下させるかどうかは証明されていないこと。 | ||
| (5) | 本剤を含むヌクレオシド系逆転写酵素阻害薬の単独投与又はこれらの併用療法により、重篤な乳酸アシドーシス及び重度の脂肪沈着による肝腫大(脂肪肝)が、女性に多く報告されているので、乳酸アシドーシス又は肝細胞毒性が疑われる臨床症状又は検査値異常(アミノトランスフェラーゼの急激な上昇等)が認められた場合には、本剤の投与を一時中止すること。これら肝疾患を発現する危険因子を有する患者においては注意すること。 | ||
| 3. | 相互作用 | ||||||
[併用注意](併用に注意すること)
| |||||||
| また、本剤の相互作用に関し、次の知見が得られている。 | |||||||
| (1) | ヒト肝ミクロソームを用いたin vitro試験において、本剤の臨床使用量では、チトクロームP450(2C9、2D6、3A4)による代謝の抑制作用は認めていない。また、肝酵素誘導作用も認められていない。従って、本剤とHIVプロテアーゼ阻害薬又はP450により代謝される薬剤との間に臨床的意義のある相互作用が起こる可能性は低いと考えられる。 | ||||||
| (2) | 本剤及びジドブジンはグルクロニルトランスフェラーゼによる共通の代謝経路を有している。海外のHIV感染症患者15名を対象としたクロスオーバー試験において、本剤600mgとラミブジン150mgの併用、ジドブジン300mgの併用又はこれら2剤併用のそれぞれの群において、臨床的に重要な本剤の薬物動態の変化は認めなかった。また、ラミブジン併用時にラミブジンの全身曝露量(AUC)は15%減少し、ジドブジン併用時ではAUCが10%増加したが、臨床的に重要な変化ではないことが示されている。 | ||||||
| 4. | 副作用 | |||||||||||||
| <国内における臨床成績> | ||||||||||||||
| 本剤を用いた日本人における臨床成績は得られていない。 | ||||||||||||||
| <海外における臨床成績> | ||||||||||||||
| 本剤によるHIV感染症の治療中に報告された有害事象は成人及び小児で同様であった。これら事象の多くは、本剤又はHIV感染症の治療中に使用された多種の薬剤に起因する症状か、あるいはHIV感染症の進展に伴い発現したものか明らかでない。またコントロールされた臨床試験において、本剤に関連する臨床検査値異常の発現はまれであり、本剤投与群と対照群との間に発現率に差は認められなかった。 | ||||||||||||||
| (1) | 重大な副作用 | |||||||||||||
| 過敏症: | ||||||||||||||
| 1) | 海外における臨床試験において、本剤投与患者の約3%に過敏症の発現を認めており、まれに致死的となることが報告されている。 | |||||||||||||
| 2) | 過敏症は、通常、本剤による治療開始6週以内(平均11日)に発現する。 | |||||||||||||
| 3) | 過敏症の特徴は多臓器及び全身に症状を認めることである。通常、認められる徴候又は症状は発熱、胃腸症状(嘔気、嘔吐、下痢、腹痛)、皮疹、嗜眠及び倦怠感である。他に、筋痛、関節痛、浮腫、息切れ、頭痛、感覚異常が認められる。なお、皮疹は多様であり、通常、斑状丘疹性皮疹又は蕁麻疹として認められるが、認めないこともある。 | |||||||||||||
| 4) | 身体所見では、リンパ節腫脹、ときに粘膜障害(結膜炎、口腔潰瘍)及び血圧低下が認められる。過敏症の発現に伴い、肝酵素値の上昇、CPK上昇、クレアチニン上昇及びリンパ球減少等の臨床検査値異常を認めることがある。 | |||||||||||||
| 5) | 過敏症に関連した腎不全及びアナフィラキシーが報告されている。 | |||||||||||||
| 6) | 過敏症に関連する症状は、本剤の投与継続により悪化し、通常、本剤の投与中止により回復する。 | |||||||||||||
| 7) | 本剤の過敏症発現後の再投与により、症状の再発が数時間以内に認められる。これは初回よりさらに重篤であり、生命を脅かす程度の血圧低下の発現及び死に至る可能性がある。従って、過敏症が発現した場合は、本剤の投与を中止し、決して再投与しないこと。 | |||||||||||||
| 8) | 本剤に対する過敏症の発現及びその重篤度を予測する危険因子は特定されていない。 | |||||||||||||
| (2) | その他の副作用 | |||||||||||||
本剤の投与により、次のような症状が認められている。通常、これらの症状は一過性であり、本剤による治療の規制因子とはならず、その多くは軽度又は中等度である。
| ||||||||||||||
| 5. | 高齢者への投与 |
| 本剤の高齢者における薬物動態は研究されていない。本剤の投与に際しては、患者の肝、腎及び心機能の低下、合併症、併用薬等を十分考慮すること。 |
| 6. | 妊婦、産婦、授乳婦等への投与 | |
| (1) | 妊婦又は妊娠している可能性のある婦人には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。[ヒト妊婦における本剤の安全性は確立されていない。動物において、本剤又はその代謝物は胎盤通過性であることが示されている。また、動物(ラットのみ)において、本剤の500mg/kg/日又はそれ以上の投与量(ヒト全身曝露量(AUC)の32〜35倍)で、胚又は胎児に対する毒性、すなわち、胎児の浮腫、変形及び奇形、体重減少、死産の増加が認められたとの報告がある。分娩前後において毒性を示さなかった量は160mg/kg/日(ヒト全身曝露量(AUC)の約10倍)であった。これらの異常はウサギでは認められていない。なお、動物における生殖毒性試験結果が、常にヒトに対する影響を予測できるものではない。] | |
| (2) | 本剤投与中は授乳を中止させることが望ましい。[本剤の乳児(3ヵ月以下)における安全性のデータはない。なお、ラットにおいて本剤及びその代謝物が乳汁中に移行することが報告されており、ヒトにおいても乳汁中に移行することが予想される。また、ある医療専門家は、HIVの乳児への移行を避けるため、あらゆる状況下においてHIVに感染した女性は授乳すべきでないことを推奨している。] | |
| 7. | 小児等への投与 |
| 3ヵ月以下の乳児に対する安全性は確立していない。[3ヵ月以下の乳児の安全性のデータはない。] |
| 8. | 過量投与 |
| 徴候、症状:臨床試験において本剤1回1200mg又は1日1800mgまで患者に投与され、予測できない副作用は報告されていない。なお、過量に投与された時の影響は知られていない。 処置:本剤による副作用(「副作用」の項参照)の発現を注意深く観察し、必要に応じ、一般的な支持療法を行うこと。なお、本剤が血液透析又は腹膜透析で除去されるかどうかは明らかでない。 |
| 9. | 適用上の注意 |
| 薬剤交付時:PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。(PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することが報告されている。) |
| 10. | その他の注意 | |
| (1) | 現時点において、本剤が車の運転又は機械の操作に影響を与えることを示唆する報告はない。 | |
| (2) | ラットにおける生殖試験では、本剤500mg/kgまでの投与量において、雌雄ともに受精率に影響は認められていない。 | |
| (3) | 細菌を用いた試験では変異原性を認めなかったが、ヒトリンパ球を用いたin vitro染色体異常試験、マウスリンフォーマ試験及び in vivo小核試験では陽性を認めた。これらの結果は、in vivo及びin vitroにおいて、本剤の高濃度を用いた場合に弱い染色体異常誘発効果を有することを示している。しかし、動物において腫瘍発現の危険性を示す報告はない。従って、ヒトに対する潜在的危険性と治療上の有益性を十分に検討すること。なお、これらは他のヌクレオシド系逆転写酵素阻害薬にて知られている知見と同様である。 | |
| (4) | 動物を用いた毒性試験では、臨床的に関連する知見は得られていない。 | |
| 【薬物動態】 <外国人における成績> | ||
| 1. | 吸収2),3) | |
| HIV感染症患者(n=12)を対象に本剤100、300、600、900、1200mgを単回経口投与した場合、Cmax及びAUC∞は投与量に依存して上昇した。未変化体の血漿中濃度は投与約1.5時間後に最高濃度に達し、消失半減期は約1.5時間であった。 | ||
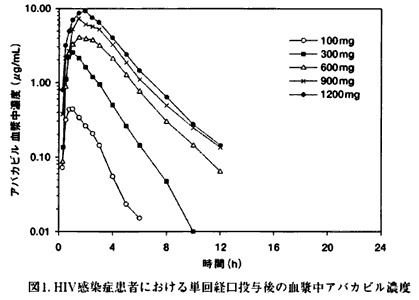
| ||
| 一方、HIV感染症患者(n=20)を対象に本剤300mgを1日2回投与した場合の定常状態におけるCmaxは約3μg/mL、12時間までのAUCは約6μg・h/mLであった。また、生物学的利用率は約83%であった。 | ||
| 2. | 分布3)(社内資料) | |
| HIV感染症患者(n=6)を対象にアバカビルを150mg静脈内投与したときの見かけの分布容積は約0.86L/kgであり、広く組織に分布することが示唆された。 本薬は10μg/mLまでの添加濃度範囲で、ヒト血漿タンパク結合率は49%と一定であった。また、血液及び血漿中放射能濃度が同じであったことから、本薬は血球に直ちに分布することが示された。 HIV感染症患者におけるアバカビルの脳脊髄液(CSF)への移行は良好で、血漿中AUCに対するCSF中AUCの比は30〜44%であった。本剤600mg1日2回投与時の最高濃度の実測値はIC50(0.08μg/mLあるいは0.26μM)の9倍であった。 | ||
| 3. | 代謝3)(社内資料) | |
| ヒトにおける主要代謝物は、5’‐カルボン酸体及び5’‐グルクロン酸抱合体であった。ヒト肝由来試料を用いたin vitro試験から、本薬は肝可溶性画分により酸化的代謝を受け5’‐カルボン酸体を生成したが、肝ミクロソーム画分では本薬の酸化的代謝は起らなかった。本薬の酸化代謝にはチトクロームP‐450ではなく、アルコールデヒドロゲナーゼ/アルデヒドデヒドロゲナーゼ系が関与していた。なお、これらの代謝物には抗ウイルス活性はなかった。 さらに、ヒト肝ミクロゾームを用いたin vitro試験において、臨床使用量での血漿中濃度ではチトクロームP‐450分子種CYP 2D6,2C9及び3A4を阻害しないことが示唆された。 | ||
| 4. | 排泄3) | |
| HIV感染症患者(n=6)を対象に14C標識アバカビル600mgを単回経口投与後、薬物体内動態を検討した。総放射能の約99%が排泄され、主な排泄経路は尿(約83%)であり、糞中には約16%排泄された。尿中に排泄された放射能の約1%は未変化体であり、約30%が5’‐カルボン酸体、約36%が5’‐グルクロン酸抱合体であった。 | ||
| 5. | 薬物相互作用 | |
| 本薬の主要代謝酵素であるアルコールデヒドロゲナーゼ/アルデヒドデヒドロゲナーゼ系への阻害効果をin vitro試験において検討した結果、本薬自身、これらの酵素を阻害しなかった(社内資料)。 ヒト肝スライスを用いたin vitro試験において、HIVプロテアーゼ阻害薬であるアンプレナビルは本薬の代謝を阻害しなかった(社内資料)。 HIV感染症患者(n=25)を対象に本剤600mgをエタノール0.7g/kgと併用して単回投与した場合、本剤のAUC∞の上昇及び t1/2の延長がみられたが臨床上重要なものではなかった。また、本剤はエタノールの薬物動態に影響を示さなかった(社内資料)。 HIV感染症患者(n=15)を対象に本剤600mgとジドブジン(300mg)及びラミブジン(150mg)のどちらか1剤あるいは両剤を併用した場合、いずれの併用においても併用薬による本剤血中濃度への影響は見られなかった。一方、本剤と併用したラミブジンのAUC∞及びCmaxは、ジドブジン併用、非併用に関わらずいずれも低下した。また、本剤と併用したジドブジンは、ラミブジン併用時及び非併用時においてAUC∞の上昇がみられたが、Cmaxは低下した。これらの変化は臨床上重要なものではなかった4)。 | ||
| 6. | 食事の影響3) | |
| HIV感染症患者(n=9)を対象に本剤300mgを食後単回投与した場合、Cmaxは空腹時単回投与と比べ低下したが、AUC∞に変化はみられなかった。 | ||
| 7. | 腎機能障害患者3) | |
| 未変化体は腎排泄が主たる排泄経路ではないため、腎機能障害患者における薬物動態は検討されていない。 | ||
| 8. | 小児(社内資料) | |
| アバカビルは経口投与後速やかにかつ良好に吸収され、成人と類似した薬物動態が認められる。3ヵ月から12歳までの小児における推奨用量は8mg/kg1日2回であり、やや高い血漿中濃度が認められるが、成人に300mg1日2回投与した時に得られる血漿中濃度に相当する。 | ||
| 【臨床成績】 <外国人における成績> | |
| 1. | 抗HIV薬による治療経験のない成人HIV感染症患者を対象とする二重盲検比較試験(試験CNAAB3003)(社内資料) CD4リンパ球数が100/mm3以上で抗HIV薬による治療経験のない成人HIV感染症患者173例を対象とした二重盲検比較試験において、本剤(300mg 1日2回)/ラミブジン(150mg 1日2回)/ジドブジン(300mg 1日2回)を併用投与した群(本剤併用群)あるいはプラセボ/ラミブジン(150mg 1日2回)/ジドブジン(300mg 1日2回)を併用投与した群(プラセボ併用群)に、それぞれ87例と86例の患者が無作為に割り付けられた。16週間の治療期間中に、血漿中HIV‐1 RNA量が検出限界(400copies/mL)以下であった患者の比率の推移を図2に示した。その結果、本剤併用群とプラセボ併用群における検出限界以下の患者の比率は治療開始8週後より差がみられ、16週後ではそれぞれ74.7%と34.9%となり、プラセボ併用群に比べて本剤併用群の方が高い抗HIV効果を示した。 |
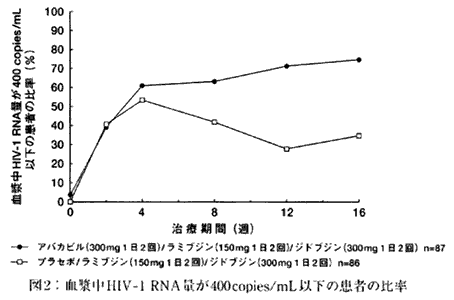
| |
| また、16週後のCD4リンパ球数の増加量(中央値)は、本剤併用群とプラセボ併用群でそれぞれ47/mm3、112/mm3であったが、両併用群とも症例毎の変化のバラツキが大きく、有意差はみられなかった。 | |
| 2. | 抗HIV薬による治療経験のある小児HIV感染症患者を対象とする二重盲検比較試験(試験CNAA3006)(社内資料) 抗HIV薬による治療経験のある生後3ヵ月〜12歳の小児HIV感染症患者205例を対象とした二重盲検比較試験において、アバカビル(8mg/kg 1日2回)/ラミブジン(4mg/kg 1日2回)/ジドブジン(180mg/m2 1日2回)を併用投与した群(アバカビル併用群)あるいはプラセボ/ラミブジン(4mg/kg 1日2回)/ジドブジン(180mg/m2 1日2回)を併用投与した群(プラセボ併用群)に、それぞれ102例と103例の患者が無作為に割り付けられた。24週間の治療期間中に、血漿中HIV‐1 RNA量が10,000copies/mL以下又は検出限界(400copies/mL)以下であった患者の比率を図3に示した。その結果、アバカビル併用群とプラセボ併用群における患者の比率は、治療開始2週後より差がみられ、24週後ではそれぞれ10,000copies/mL以下が46.1%、36.9%、検出限界以下が11.8%、1.0%であり、プラセボ併用群に比べてアバカビル併用群の方が高い抗HIV効果を示した。 |
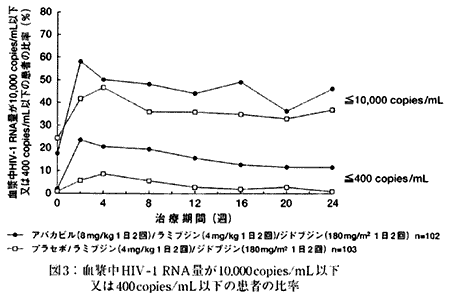
| |
| また、16週後のCD4リンパ球数の増加量(中央値)は、プラセボ併用群の9/mm3に比べてアバカビル併用群が69/mm3であり、プラセボ併用群に比べてアバカビル併用群の方が多かった。 | |
| 【薬効薬理】 | |
| 1. | 作用機序5) |
| アバカビルは細胞内で細胞性酵素によって活性代謝物のカルボビル三リン酸に変換される。カルボビル三リン酸は天然基質dGTPと競合し、ウイルスDNAに取り込まれることによって、HIV‐1逆転写酵素(RT)の活性を阻害する。取り込まれたヌクレオシド誘導体には3’‐OH基が存在しないため、DNA鎖の伸長に不可欠な5’‐3’ホスホジエステル結合の形成が阻害され、ウイルスのDNA複製が停止する。 | |
| 2. | 抗ウイルス作用(In vitro)6),7) |
| アバカビルのHIV‐1に対するIC50値はHIV‐1 IIIBに対して3.7〜5.8μM、臨床分離株に対して0.26±0.18μM(1μM=0.28μg/mL)(n=8)、HIV‐1 BaLに対して0.07〜1.0μMであった。In vitroでアンプレナビル、ネビラピン及びジドブジンとの併用によって相乗作用が認められ、ジダノシン、ラミブジン、サニルブジン及びザルシタビンとの併用によって相加作用が認められた。また、ヒト末梢血単核球から活性化リンパ球を除いた場合に、より強い抗HIV作用を示したことから、アバカビルは静止細胞でより強く抗ウイルス作用を示すものと考えられる。 | |
| 3. | 薬剤耐性3),8) |
| アバカビルに対して低感受性のHIV‐1分離株がin vitroで選択されたほか、アバカビル投与患者からも分離されている。アバカビル投与患者の分離株の解析では、アミノ酸をコードしている遺伝子の点突然変異による、逆転写酵素の65番目のアミノ酸がリジンからアルギニンへ、74番目のアミノ酸がロイシンからバリンへ、115番目のアミノ酸がチロシンからフェニルアラニンへ、また184番目のアミノ酸がメチオニンからバリンへの変異が確認された。臨床分離株では184番目のアミノ酸がメチオニンからバリンへの変異、及び74番目のアミノ酸がロイシンからバリンへの変異が頻回に観察された。アバカビルを12週間単独投与した患者17例から採取したアバカビル誘発突然変異をもつHIV‐1分離株のアバカビルに対する感受性をin vitroで検討した結果、アバカビル感受性は 1/3 であったが、アバカビル投与による遺伝子変異とアバカビル感受性の臨床的相関は判明していない。 | |
| 4. | 交差耐性8) |
| アバカビルに耐性を示す複数の逆転写酵素変異を組み込んだHIV‐1株は、in vitroでラミブジン、ジダノシン及びザルシタビンに対して交差耐性を示した。 アバカビルとHIVプロテアーゼ阻害薬とは標的酵素が異なることから、両者間に交差耐性が発生する可能性は低く、非ヌクレオシド系逆転写酵素阻害薬も逆転写酵素の結合部位が異なることから、交差耐性が発生する可能性は低いものと考えられる。 | |
| 【有効成分に関する理化学的知見】 | |
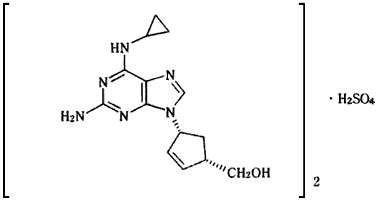
| 一般名: | 硫酸アバカビル(Abacavir Sulfate) |
| 化学名: | (−)‐{(1S,4R)‐4‐[2‐アミノ‐6‐(シクロプロピルアミノ)プリン‐9‐イル]シクロペンタ‐2‐エニル} メタノール 1/2硫酸塩 |
| 分子式: | (C14H18N6O)2・H2SO4 |
| 分子量: | 670.76 |
 | |
| 性 状: | 白色〜微黄白色の粉末である。0.1 mol/L塩酸試液に溶けやすく、水にやや溶けやすく、希水酸化ナトリウム試液にやや溶けにくい。 |
| 融 点: | 約219℃(分解) |
| 分配係数:1.20(pH 7.1〜7.3,1‐オクタノール/水) |
| 【承認条件】 | |
| 1) | 国内における薬物動態試験については、試験終了次第、可及的速やかに試験成績、解析結果を提出すること。 |
| 2) | 本剤を使用する場合は、過敏症に留意して、過敏症等の副作用が発生した場合には必ず適切な処置をとるよう、また、治療にあたっては、本剤は現在我が国における臨床試験が行われており、薬剤に関する科学的なデータを収集中であること等、患者に十分な説明を行い、インフォームド・コンセントを得るよう医師に対して要請すること。 |
| 3) | 臨床試験については、プロトコールを遵守し、定期的(6ヵ月に1回程度を目途)に試験成績を報告し、試験終了次第、可及的速やかに試験成績、解析結果を提出すること。 |
| 4) | 今後、再審査期間の終了までは、国内で使用される症例に関しては、可能な限り全投与症例を市販後調査の対象とし、患者背景、臨床効果、副作用、薬物相互作用等に関してデータの収集を行い、再審査の申請資料として提出すること。 |
| 5) | 市販後、本剤の使用実態について詳細に調査を行い、他剤との併用における本剤の安全性、有効性に関する情報収集を実施し、定期的に報告すること。 |
| 【包 装】 |
| ザイアジェン錠:100錠 (10錠×10) PTP |
| 【主要文献】 | |
| 1) | Charles, C. J., et al.:JAMA, 277(24), 1962‐1969 (1997) |
| 2) | Princy, N. K., et al.:Antimicrob. Agents Chemother., 43(3), 603‐608 (1999) |
| 3) | ザイアジェン錠米国添付文書 |
| 4) | Laurene, H. W., et al.:Antimicrob. Agents Chemother., 43(7), 1708‐1715 (1999) |
| 5) | Faletto, M. B., et al.:Antimicrob. Agents Chemother., 41(5), 1099‐1107 (1997) |
| 6) | Daluge, S. M., et al.:Antimicrob. Agents Chemother., 41(5), 1082‐1093 (1997) |
| 7) | Saavedra, J., et al.:Intersci. Conf. Antimicrob. Agents Chemother. (ICAAC) 253 (1997) |
| 8) | Tisdale, M., et al.:Antimicrob. Agents Chemother., 41(5), 1094‐1098 (1997) |
| ■ 過敏症を注意するカード | ||||||
|



| TOPに戻る |